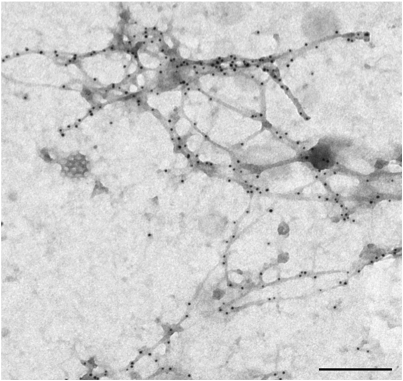
Actin cytoskeleton has been implicated at early FcεRI-mediated signaling events as well as at later steps leading to degranulation and/or cell migration (1). In initial biochemical studies aggregation of the FcεRI caused rapid decrease in the amount of filamentous (F)-actin followed by its subsequent increase exceeding the levels typical for nonactivated cells (2,3). Other studies with permeabilized mast cells where F-actin was detected by means of fluorescently labeled phalloidin confirmed transient changes in F-actin content (2,4). Interestingly, IgE binding itself has been shown to increase the content of cellular F-actin (5,6). Redistribution of actin filaments in activated mast cells is an essential step leading to such important morphological changes as formation of membrane ridges, microvilli and cell spreading (Fig. 1). F-actin outlines the cell surface lamellae, which indicates that F-actin complexes are mostly associated with the plasma membrane (3,7).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Actin microfilaments at early FceRI-mediated signaling events
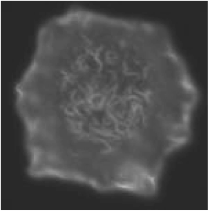
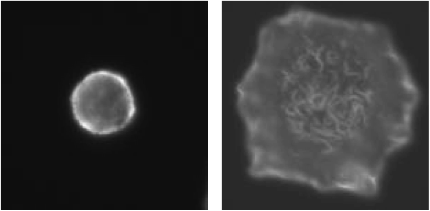
Most of the experiments aiming at better understanding of the role of F-actin in mast cell signaling utilized drugs which interfere with F-actin formation, namely latrunculin B and cytochalasins. Pretreatment of mast cells with these drugs enhanced antigen-induced tyrosine phosphorylation of the FcεRI b and g subunits, which is the earliest known biochemical event after FceRI triggering. Latrunculin B alone also caused a small increase in tyrosine phosphorylation of FceRI subunits. This suggests that microfilaments also regulate the level of receptor tyrosine phosphorylation in resting cells (4). Actin filaments are involved in the regulation of the kinetics of antigen-mediated tyrosine phosphorylation. Some data indicate that actin filaments are important in limiting the lifetime of FcεRI-Lyn interactions (8,9). However, treatment of mast cells with cytochalasin D or latrunculin A prior to FcεRI aggregation failed to cause any increase in the amount of Lyn kinase associated with FcεRI (10). Electron microscopy studies showed that oxidized (inhibited) protein tyrosine phosphatases (PTPs) are associated with plasma membrane-bound actin cytoskeleton (Fig. 2) and that their number increased after antigen triggering. This could be related to an important role of the actin-bound PTPs in regulation of FcεRI signaling (11,12).
Detailed studies ivestigating real-time kinetics of actin and FcεRI in mast cells revealed receptor movement within micron-sized membrane domains defined by actin bundles; this confinement was dynamic over length-scales of microns and time-scales of seconds. The diffusion of FcεRI complexes decreases within seconds after addition of the receptor crosslinking agent (13).
Actin microfilaments at later stages of FcεRI signaling and degranulation
Latrunculin B increased the rate and extent of FcεRI-induced degranulation, calcium response, phosphorylation of several signaling proteins (e. g. Syk, PLCg, Gab2 and LAT) and enzymatic activity of PI3K, PLCg and SHP-2 (4,14). It has been reported that FcεRI triggering leads to disassembly of cortical F-Actin ring. This has been considered an important step facilitating fusion of granules and plasma membranes in Ca2+ dependent manner (15). However, disassembly of F-actin cortex alone was not sufficient for degranulation; actin seems to play a modulatory rather than a central role at this stage (16,17). In addition, actin cytoskeleton is closely involved in determining the properties of the plasma membrane and affects the rate of fusion between secretory granules and plasma membrane (18). Reorganization of actin microfilaments is controled by Ca2+-dependent actin regulatory proteins (16,19). There seem to be other mechanisms which are calcium independent (20). One of the possible prerequisites for F-Actin disassembly is Ca2+-independent association of calmodulin with the actin cortex in activated cells and promotion of myosin II-based contraction of the membrane cytoskeleton (21,22). FcεRI triggering induced the subcellular relocation of some members of Coronin protein family. These proteins seem to cooperate with actin cytoskeleton during cell activation and contribute to reduced cortical stability and better access of secretory granules to the plasma membrane (23). The other group of proteins involved in regulation of actin dynamics consists of Ezrin/Radixin/Moesin (ERM) family proteins. These proteins mediate the anchoring of cortical F-actin to the plasma membrane (14,24). In activated mast cells, protein kinase C (PKC) is likely to participate in the regulation of morphological changes by promoting the membrane ruffling and cell spreading, as well as binding to fibronectin and chemotaxis toward antigen (25). However the F-actin response in mast cells depended on PKC when the cells were triggered through FceRI but was PKC independent when triggered through the adenosine receptor (2). Rho-related small GTPases (Rho, Rac, Cdc42) are known to regulate changes in cell shape, movement and adhesion of mammalian cells. Rac1 and RhoA have been shown to control both the secretion and actin organization in mast cells (26,27). Rho was found to regulate de novo actin polymerization (28) and cortical F-actin disassembly (19). Cdc42 seems to control distinct pathways downstream of FcεRI than those controlled by Rac1, and probably act at different stages of the secretory process by controlling distinct cytoskeletal structures; Rac1 may regulate the access or the transport of cytoplasmic granules to the plasma membrane, while CDC42 may influence critical events in secretion by controlling the distribution of the secretory granules within the cells (29). FcεRI triggering caused an increase in activity of Rac(1, 2, 3) while decreasing the activity of RhoA, which was shown to regulate actin polymerization at later time intervals after triggering; this process involves adaptor protein NTAL (7). PI3K was shown to regulate changes in F-Actin during mast cell chemotaxis upstream of Rac (30). Several other regulators and/or effectors of Rho GTPases were shown to be important factors in mast cell function, e. g. p21-activated kinase 1 (Pak1) (5), swich-associated protein 70 (SWAP-70) (31), Wiskott-Aldrich syndrome protein-interacting protein (WIP) (32), and Bruton´s tyrosin kinase (Btk) (30).
References:
| 1. | Dráber P, Sulimenko V, & Dráberová E. (2012) Front. Immunol. 3, 130. |
| 2. | Apgar J R. (1994) Mol. Biol. Cell 5, 313-322. |
| 3. | Pfeiffer J R, Seagrave J C, Davis B H, Deanin G G, & Oliver J M. (1985) J. Cell Biol. 101, 2145-2155. |
| 4. | Frigeri L, & Apgar J R. (1999) J. Immunol. 162, 2243-2250. |
| 5. | Allen J D, Jaffer Z M, Park S-J, Burgin S, Hofmann C, Sells M A, Chen S, Derr-Yellin E, Michels E G, McDaniel A, Bessler W K, Ingram D A, Atkinson S J, Travers J B, Chernoff J, & Clapp D W. (2009) Blood 113, 2695-2705. |
| 6. | Oka T, Hori M, Tanaka A, Matsuda H, Karaki H, & Ozaki H. (2004) Am. J. Physiol. Cell Physiol. 286, C256-C263. |
| 7. | Tůmová M, Koffer A, Šimíček M, Dráberová L, & Dráber P. (2010) Eur. J. Immunol. 40, 3235-3245. |
| 8. | Holowka D, Sheets E D, & Baird B. (2000) J. Cell Sci. 113, 1009-1019. |
| 9. | Wilson B S, Pfeiffer J R, & Oliver J M. (2000) J. Cell Biol. 149, 1131-1142. |
| 10. | Torigoe C, Song J, Barisas B G, & Metzger H. (2004) Mol. Immunol. 41, 817-829. |
| 11. | Bugajev V, Bambousková M, Dráberová L, & Dráber P. (2010) FEBS Lett. 584, 4949-4955. |
| 12. | Heneberg P, Dráberová L, Bambousková M, Pompach P, & Dráber P. (2010) J. Biol. Chem. 285, 12787-12802. |
| 13. | Andrews N L, Lidge K A, Pfeiffer J R, Burns A R, Wilson B S, Oliver J M, & Lidge D S. (2008) Nat. Cell Biol. 10, 955-963. |
| 14. | Tolarová H, Dráberová L, Heneberg P, & Dráber P. (2004) Eur. J. Immunol. 34, 1627-1636. |
| 15. | Nishida K, Yamasaki S, Ito Y, Kabu K, Hattori K, Tezuka T, Nishizumi H, Kitamura D, Goitsuka R, Geha R S, Yamamoto T, Yagi T, & Hirano T. (2005) J. Cell Biol. 170, 115-126. |
| 16. | Koffer A, Tatham P E, & Gomperts B D. (1990) J. Cell Biol. 111, 919-927. |
| 17. | Pendleton A, & Koffer A. (2001) Cell Motil. Cytoskelet. 48, 37-51. |
| 18. | Deng Z, Zink T, Chen H-y, Walters D, Liu F-t, & Liu G-y. (2009) Biophys. J. 96, 1629-1639. |
| 19. | Sullivan R, Price L S, & Koffer A. (1999) J. Biol. Chem. 274, 38140-38146. |
| 20. | Guzmán R E, Bolanos P, Delgado A, Rojas H, DiPolo R, Caputo C, & Jaffe E H. (2007) Pflueg. Arch. Eur. J. Physiol. 454, 131-141. |
| 21. | Sullivan R, Burnham M, Torok K, & Koffer A. (2000) Cell Calcium 28, 33-46. |
| 22. | Psatha M, Koffer A, Erent M, Moss S E, & Bolsover S. (2004) Cell Calcium 36, 51-59. |
| 23. | Foger N, Jenckel A, Orinska Z, Lee K-H, Chan A C, & Bulfone-Paus S. (2011) J. Exp. Med. 208, 1777-1787. |
| 24. | Staser K, Shew M A, Michels E G, Mwanthi M M, Yang F-C, Wade Clapp D, & Park S-J. (2012) Exp. Hematol. (in press). |
| 25. | Yanase Y, Hide I, Mihara S, Shirai Y, Saito N, Nakata Y, Hide M, & Sakai N. (2010) Immunol. Cell Biol. 89, 149-159. |
| 26. | Norman J C, Price L S, Ridley A J, & Koffer A. (1996) Mol. Biol. Cell 7, 1429-1442. |
| 27. | Price L S, Norman J C, Ridley A J, & Koffer A. (1995) Curr. Biol. 5, 68-73. |
| 28. | Norman J C, Price L S, Ridley A J, Hall A, & Koffer A. (1994) J. Cell Biol. 126, 1005-1015. |
| 29. | Guillemot J C, Montcourrier P, Vivier E, Davoust J, & Chavrier P. (1997) J. Cell Sci. 110, 2215-2225. |
| 30. | Kuehn H S, Radinger M, Brown J M, Ali K, Vanhaesebroeck B, Beaven M A, Metcalfe D D, & Gilfillan A M. (2010) J. Cell Sci. 123, 2576-2585. |
| 31. | Sivalenka R R, & Jessberger R. (2004) Mol. Cell. Biol. 24, 10277-10288. |
| 32. | Kettner A, Kumar L, Antón I M, Sasahara Y, de la Fuente M, Pivniouk V I, Falet H, Hartwig J H, & Geha R S. (2004) J. Exp. Med. 199, 357-368. |